Setting up your antiSMASH job#
This page describes all relevant steps to get to interpretable antiSMASH output using the web server. It covers everything from the correct input data to what all settings and parameters mean. Here is the latest stable bacterial version of antiSMASH available of which a screenshot is provided below:
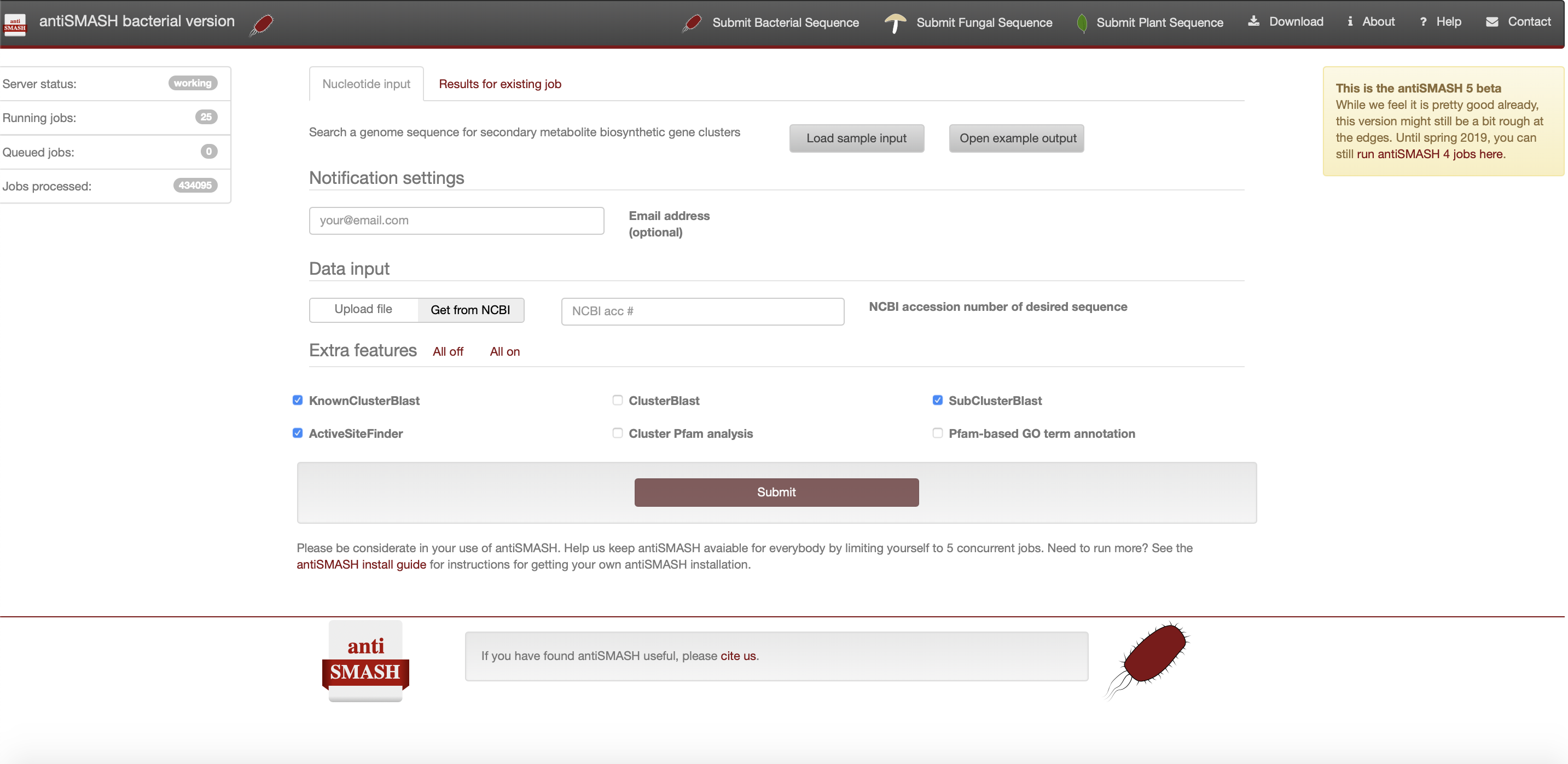
Registering your antiSMASH job#
Please make sure that if you have fungal or plant sequence data that you click on the appropriate tab in the top bar as some settings are dedicated to either bacterial or fungal data and to upload plant sequences you are redirected to PlantiSmash.
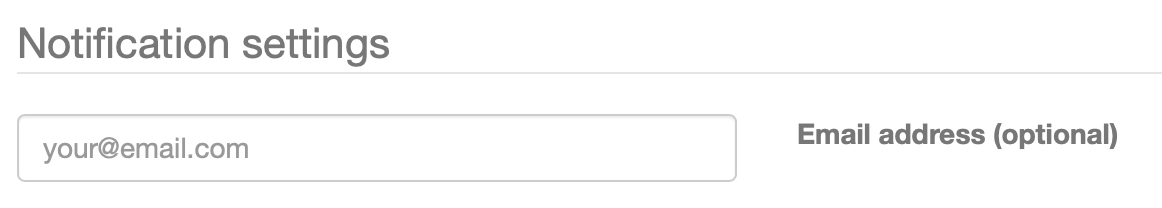
In case you would like to get an email alert upon completion of your job you can optionally provide your email address in the "Notification settings" panel. You will then receive an email with a link to your completed job once it finished okay or if any error would have unfortunately occurred. Please note that if you did not provide an email address, you should bookmark the link to the job submission page, as otherwise you will not be able to access the results anymore.
antiSMASH input data#
The ideal input for antiSMASH is an annotated nucleotide file in Genbank format or EMBL format. You can either upload a local GenBank/EMBL file manually, or simply enter the GenBank/RefSeq accession number of your sequence for antiSMASH to upload it. You can toggle the option in the "Data input" panel where default is the accession number entry where antiSMASH will attempt to download the data directly from NCBI.

In case no gene annotations are available for your sequence, we recommend running your sequence through an annotation pipeline like RAST to obtain GBK/EMBL files with high-quality annotations. Alternatively, you can provide a FASTA file containing a single sequence. In that case, antiSMASH will generate a preliminary annotation using Prodigal, and use that to run the rest of the analysis. You can also provide gene annotations in GFF3 format.
In any case, it is very important that input files are properly formatted. If you are creating your GBK/EMBL/FASTA file manually, be sure to do so in a plain text editor like Notepad or Emacs, and saving your files as "All files (.)", ending with the correct extension (for example ".fasta", ".gbk", or ".embl".
antiSMASH default and extra features#
Before pressing the submission button, you will have to indicate which antiSMASH features you like to run.
Default antiSMASH features#
The following options are run by default if you do not toggle them off as can be seen in the screenshot from the "Extra features" panel:
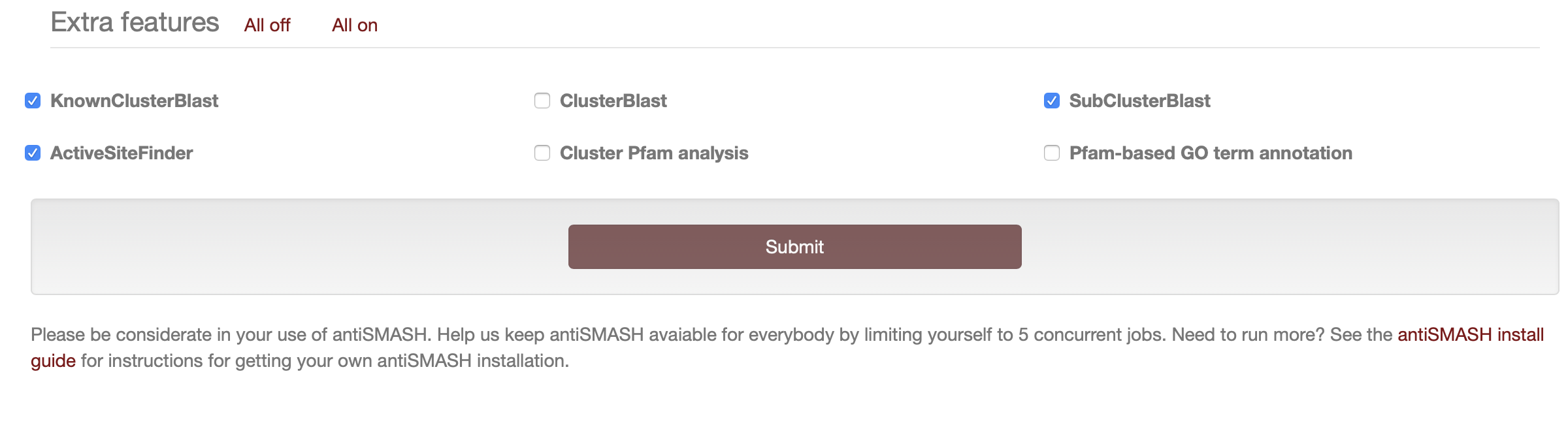
KnownClusterBlast analysis: the identified clusters are searched against the MIBiG repository. MIBiG is a hand curated data collection of biosynthetic gene clusters, which have been experimentally characterized.
Subcluster Blast analysis: the identified clusters are searched against a database containing operons involved in the biosynthesis of common secondary metabolite building blocks (e.g. the biosynthesis of non-proteinogenic amino acids).
The following antiSMASH features always run in the background:
smCOG analysis: the analysis of secondary metabolism gene families (smCOGs) attempts to allocate each gene in the detected gene clusters to a secondary metabolism-specific gene family using profile hidden Markov models specific for the conserved sequence region characteristic of this family. In other words, each gene of the cluster is compared to a database of clusters of orthologous groups of proteins involved in secondary metabolism. Additionally, a phylogenetic tree is constructed of each gene together with the (max. 100) sequences of the smCOG seed alignment. This information can then be used to provide an annotation of the putative function of the gene products. smCOG analysis results can be used for functional prediction and phylogenetic analysis of genes.
Active site finder: active sites of several highly conserved biosynthetic enzymes are detected and variations of the active sites are reported.
TTA codon detection: high-GC containing bacterial sequences (by default in antiSMASH >65%), for example from Streptomycetes, contain the rare Leu-codon “TTA” as a mean for post-transcriptional regulation by limiting/controlling the amount of TTA-tNRA in the cell. This type of regulation is commonly found in secondary metabolite BGCs. This feature will annotate such TTA codons in the identified BGCs.
Advanced antiSMASH features#
As can be seen in the screenshot above, all features can be easily toggled off or on in one click on the top of the "Extra features" panel. The following three features are off by default and are considered to be useful for advanced users or in case interesting biosynthetic gene clusters were found that warrant further detailed analysis.
ClusterBlast analysis: the identified clusters are searched against a comprehensive gene cluster database and similar clusters are identified. The algorithm used here is inspired by MultiGeneBlast. It runs BlastP using each amino acid sequence from a detected gene cluster as a query on a large database of predicted protein sequences from secondary metabolite biosynthetic gene clusters, and pools the results to identify the gene clusters that are most homologous to the gene cluster that was detected in your query nucleotide sequence. Enabling this option will increase the runtime of a submission.
Cluster Pfam analysis: each gene product encoded in the detected BGCs is analyzed against the PFAM database. Hits are annotated in the final Genbank/EMBL files that can be downloaded after the analysis is finished. Please note that these results are not displayed on the antiSMASH HTML results page but they are present in the results genbank file that can be downloaded. Enabling this option will increase the runtime of a submission.
Pfam-based GO term annotation: this option annotates the results of the Cluster Pfam analysis, described above, with GO term annotations. These results are displayed within the Pfam domains section of the antiSMASH HTML results, by emphasising the borders of the relevant domains. See Understanding the output for instructions on how to download the results.
FungiSMASH#
The fungal version of antiSMASH, FungiSMASH, has some small modifications to better support detection and analysis.